
Life’s origins lie more than 3800 million years ago. At some point since, a common ancestor to all living organisms existed, and for thousands of millions of years, lineages have been born, split, and gone extinct. The relationships of these lineages can be visualised as a tree: some lineages are more closely related to each other (split more recently) than they are to everything else.
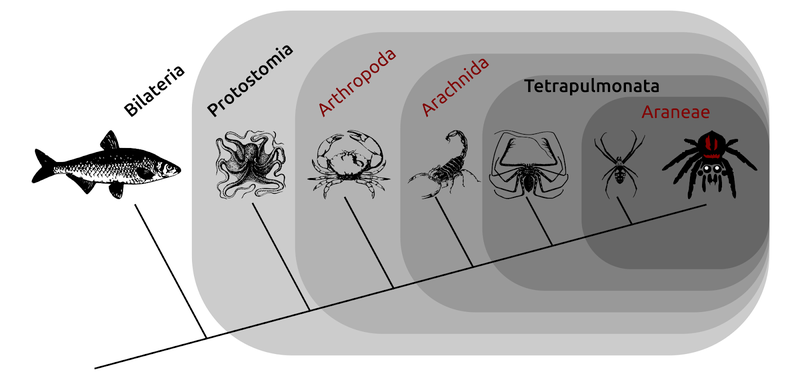
For example, it may not be immediately obvious, but animals with backbones are more closely related to sea urchins and other organisms with five-fold symmetry than they are to all other animals. Another example of a tree is shown in the figure below, which is a family tree of some (but not all) of the arachnids and related animals with twofold symmetry. In this case, for example, spiders (Araneae) diversified most recently, and all share the ability to spin silk. They share with other arachnids such as the whip spiders, the presence of four lungs for breathing (hence Tetrapulmonata - "four lungs"), so that was the split before the spiders diversified. The arachnids more widely share four walking leg pairs, and all share a more recent common ancestor (which probably had four legs too...) than the shared ancestor between arachnids and, say, crustaceans (including crabs, lobsters, and shrimp). Phylogenetic reconstruction is the process by which we try and deduce these evolutionary relationships. So far so good.
There are a few ways we can reconstruct phylogenies. Since the advent of computers, we have codified anatomy (e.g. how many legs does a creature have? How many eyes?) and then conducted a search. For example, a long standing approach is termed parsimony - we try lots of different tree arrangements, and assume that the arrangement that implies the smallest number of character changes is the best (we may group all vertebrates with four legs together say, rather than assuming that multiple lineages evolved a four-legged form).
In recent years, computers have become powerful enough to use probabilistic approaches. A common one is a form of Bayesian analysis - if you have a tree shape, our coded morphology, and a model of evolution, you can work out the probability of any given tree. In order to use this approach to deduce an evolutionary tree that incorporates uncertainty, we use a Markov chain Monte Carlo (MCMC chain) - a computationally expensive approach, which you can read about in gory (but hopefully approachable) detail in my PALAEONTOLOGY[ONLINE] article.
Along with the rise of probabilistic approaches, recent decades have seen the increasing use of DNA to build phylogenies. If you get the genome of two animals, the more closely related they are, the more similar they will expect their DNA to be. This has revolutionised our view of the relationships of many forms of life.
There is a sticking point in all of this, however. And that is fossils. Something like 99.9% of species to have ever lived are now extinct. For almost all extinct lineages, it is impossible to get their DNA as it decays with time. For dead things we’re stuck with morphology. But how best to reconstruct phylogenies from morphology, which doesn’t tend to evolve in quite so predictable a way as DNA? Well, that brings us to where we are today. There has been a recent slew of papers that have used a range of models to generate “morphological” data onto a tree, use Bayesian and parsimony to deduce a tree using that data, and then compare the deduced tree to the true tree. Given we can never be sure of the true tree in the real world, this computer-based approach is very attractive. I, along with colleagues here in Manchester and elsewhere, recently created and published a new model that creates character data and trees at the same time, using a selection-based model (as opposed to the stochastic models that have been used in this area to date).
This is lovely - we can now use the model to assess Bayesian and parsimony approaches, and to quantify (for example) the impact that including fossils has on our ability to build the true tree! And therein, as the Bard would tell us, lies the rub. We can generate so much data that the rate limiting step in our work has become running the computationally expensive Bayesian analyses. Indeed, as the COVID-19 induced lockdown hit us, my colleagues (at University of Oxford and Yale University) and I were looking down the barrel of running Bayesian phylogenies for the next three - six months on a workstation to get the initial results for once such simulation project. It is here that Research IT have come to our rescue, providing us access to the Condor Pool - a facility made up of dedicated processor cores or on-demand (spot reserved) Amazon Web services (AWS) Cloud instances.
The Research IT team built MrBayes - our software of choice - on the HTCondor pool allowing us to transfer the software to any Linux node on the system, and provided an exemplar submission script. They also provided remote access to HTCondor, which is particularly important when working out of your spare room. With this head start, and an afternoon of playing with shell scripts, we were able to get a job up and running. This allowed us to run between 300 and 600 analyses at any given time, 24 hours a day. Months of analysis shrunk in an instant to a fortnight for the first tranche of data. In fact, we were so overjoyed that we set another 7500 analyses running to celebrate. That’s also finished now.
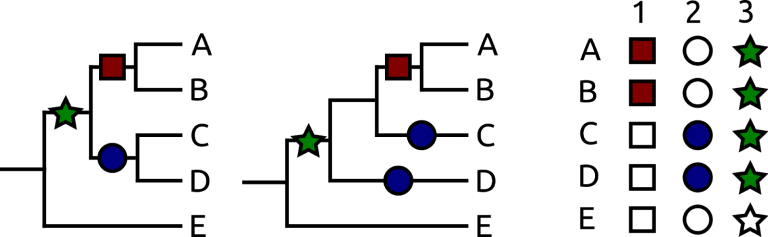
Two possible trees of five species, A–E. These have characters represented as empty and filled shapes. Assuming that empty is the original condition for all, the left tree requires one fewer state change than that on the right, and as such is more parsimonious.
So a key question is what difference does this make to our research? Well, in what is a currently fast-moving area in the world of palaeontology / phylogenetics, it has allowed our writing process to be the rate limiting step for this study, rather than the analysis time. If we get scooped, it’s our own fault. We have more replicates than we thought would have been possible at the launch of our project, strengthening our confidence in our results. Ultimately HTCondor gives us the freedom with future projects to be more ambitious when it comes to our research programme. That, right there, is a wonderful place to be.